Thoracic Aortic Aneurysm
A High Index of Clinical Suspicion
Aortic aneurysm is a common aortic pathology requiring prompt surgical intervention. The symptoms associated with an aortic aneurysm can often be subtle, including those related to aneurysm expansion (e.g. chest pain or breathlessness) and compression of adjacent structures (e.g. hoarseness or stridor). Unfortunately, many of patients may be undiagnosed or even misdiagnosed, until they present with catastrophic complications, such as acute aortic dissection, aortic rupture, cardiovascular collapse, malperfusion and/ or death. This is why ‘aneurysm’ has been perceived as the ‘Great Masquerader’ in the surgical literature (Figure 1). As the thoracic aortic aneurysm enlarges, the aortic wall tension increases. If the blood pressure is too high, the aortic aneurysm may burst or dissect, which often leads to death. We, as clinicians, ought to maintain a high index of clinical suspicion for patients with a possible aortic aneurysm.
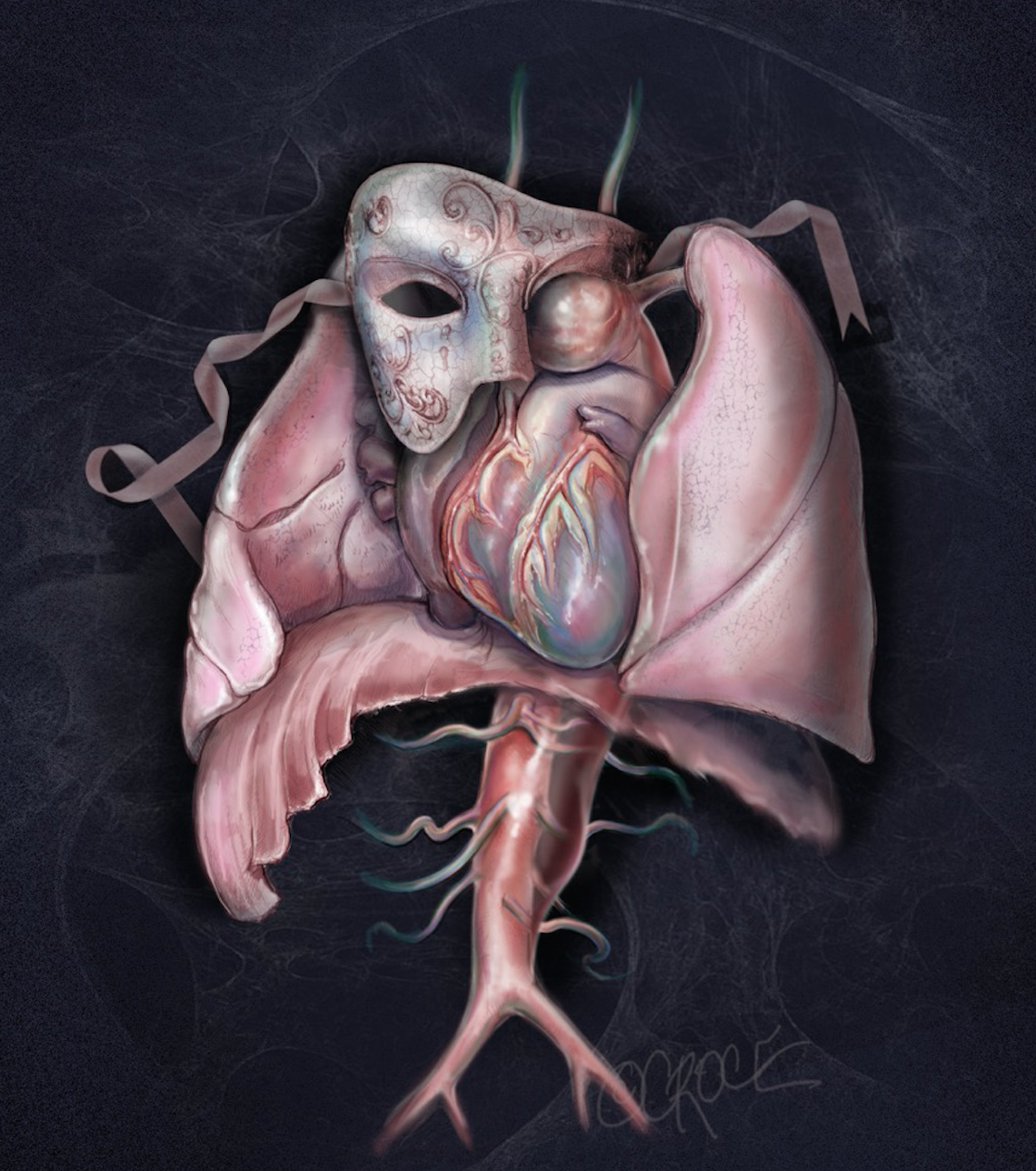
Figure 1. Thoracic Aortic Aneurysm – the ‘Great Masquerader’
Thoracic Aortic Aneurysm is a Genetic Condition
Thoracic aortic aneurysm is a genetic condition. Marfan syndrome is just the tip of the genetic iceberg. One of the simplest screening test that we do during our initial consultation is the ‘Thumb and Palm’ sign for Marfan syndrome. If the patient’s thumb can go all the way across his/her fully extended palm, it may mean that the patient’ long bones are extensive and joins are lax, a sign of connective tissue condition that may be related to Marfan syndrome.
Bicuspid aortic valve (BAV) is the most common congenital heart condition, affecting 2% of the general population. BAV is a clinically mixed disorder with a high rate of surgically relevant aortic valve and ascending aortic complications, which occur in over 35% of those affected. BAV may degenerate early and the valve may become narrowed or leaky (Figure 2).
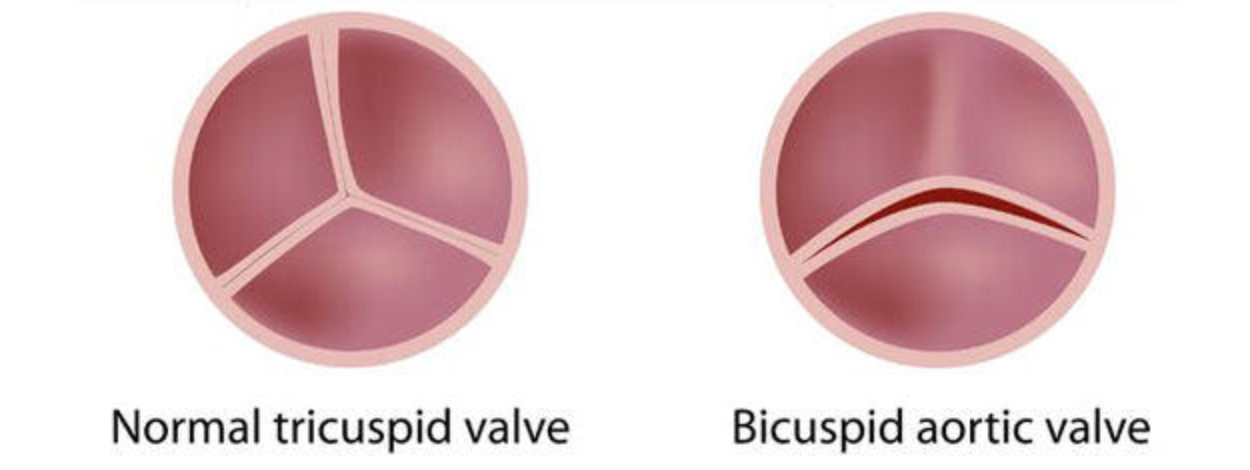
Figure 2. Bicuspid Aortic Valve is Associated with Thoracic Aortic Aneurysm
As such, BAV confers a greater burden of disease than all other congenital heart diseases combined. Familial clustering and genetic studies have established that BAV has a heritable trait, with approximately 10% prevalence amongst first-degree relatives, and up to 25% in families with more than one affected family member. Thoracic aortic aneurysm (TAA) is common in BAV, reported in up to 50 – 60% of affected individuals, predisposing to aortic dissection/rupture, the most feared complication in this relatively young population. Indeed, BAV conveys an 8-fold increased risk of aortic dissection and over a 25-year period, the risk for aneurysm formation is 26% and for aortic surgery is 25%3, further highlighting its clinical importance.
According to the Class I recommendation by the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) guidelines:
1. First-degree relatives of patients with a BAV and/or a familial form of TAA and dissection should be evaluated for the presence of a BAV and asymptomatic thoracic aortic disease.
2. All patients with a BAV should have both the aortic root and ascending thoracic aorta evaluated for evidence of aneurysm.
How to Diagnose Thoracic Aortic Aneurysm?
Initial assessment is transthoracic echocardiography to evaluate proximal aorta and valvular pathology. To visualize distal ascending aorta, aortic arch and descending thoracic aorta, computed tomography (CT) Thoracic
Aortogram is the most commonly used imaging modality for the diagnosis of TAA. It provides information regarding the size, location, and extent of the disease. The other imaging techniques include: CXR, MRI, TOE and aortography. We can perform detailed analysis, such as 3-D printing, 4-D flow MRI and Computation Fluid Dynamic Analysis to analyze aortic wall tension and sheer stress in order to monitor aortic behavior more closely (Video 2).
What are the Indicators for Surgery?
The timing for surgery is largely based on symptoms and the size of the aneurysm. In addition, surgery should be considered earlier if patients have a connective tissue disorder, such as Marfan syndrome and BAV.
CLASS I RECOMMENDATIONS
1. Patients with symptoms suggestive of expansion of a TAA should be evaluated for prompt surgical intervention unless life expectancy from comorbid conditions is limited or quality of life is substantially impaired
2. Asymptomatic patients with TAA, who are otherwise suitable candidates and for whom the ascending aortic aneurysm reaches 5.5cm should be evaluated for surgical repair.
3. Patients with connective tissue disorders (Marfan syndrome, BAV, vascular Ehlers-Danlos syndrome, Turner syndrome, or familial thoracic aortic aneurysm and dissection) should undergo elective operation at smaller diameters (4.0 to 5.0 cm) to avoid acute dissection or rupture
4. Patients with a growth rate of > 0.5 cm/year in an aorta that is < 5.5 cm in diameter should be considered for operation.
Royal Prince Alfred Hospital has the long-standing tradition of a subspecialised team approach for complex clinical conditions. There is a dedicated aortic team, formed by cardiac surgeons, cardiologists, radiologists, anaesthetists and physiotherapists who work closely together and manage patients with complex aortic conditions. Patients are discussed and looked after in this cohesive team environment. This multidisciplinary team approach offers the best treatment option (open, endovascular vs. hybrid) for each individual patient. For consultation, please contact Professor Tristan D. Yan's rooms.
Book Appointment

Mini-Bentall Procedure
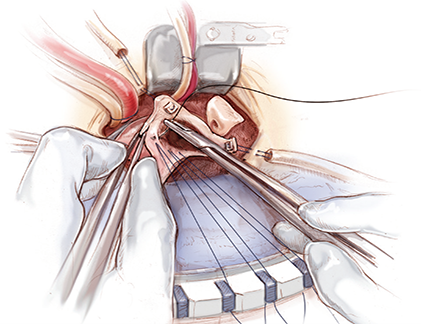
Mini-Ascending Aortic Surgery
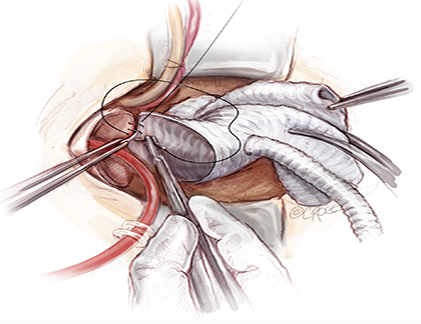
Mini-Arch Surgery